References:
- Lecturer (Cainglet, delos Reyes)
The assessment for a newborn does not follow the usual cephalocaudal direction. It is used starting in preschoolers (4 to 6 years old). For younger children, the chest is the first target, before proceeding with the head. This discussion utilizes a assessment-based approach, categorizing normal and abnormal findings based on the body system being assessed. Particularly lengthy discussions on abnormalities will be in their own section.
Chest Assessment
The chest should not show retractions, and have clear symmetrical breath sounds throughout all lung fields.
Abnormalities
- Retractions: visual inspection for retractions that may indicate respiratory distress.
- Breath Sounds: auscultate the lungs for breath sounds.
- If absent, the lungs are not expanding; collapsed lungs or atelectasis.
- Wheezes, especially upon expiration, may indicate a lower respiratory tract obstruction.
- Stridor, high-pitched adventitious sounds upon inspiration indicates an upper respiratory tract obstruction.
- Rales or crackles are rattling, bubbling, or clicking sounds. This is commonly found in fluid or secretory obstruction in the lungs.
- Witch Milk: colorless or transparent fluid expressed by the neonatal mammary glands caused by maternal hormones.
Head Assessment
Two important landmarks of the hear are the fontanels. There are six in total: the anterior fontanel, the posterior fontanel, two sphenoid (anterolateral) fontanel, and two mastoid (posterolateral) fontanel.
- The anterior fontanel is diamond-shaped and 3-4 cm long and 2-3 cm wide. If the fontanel is larger than 5 cm, it may indicate hydrocephaly or cretinism. It closes within 12 to 18 months, as early as 9 months. Earlier closure is termed “Craniostenosis”.
- The posterior fontanel is triangle-shaped and measures 2 cm long and 1 cm wide. It closes within 2 to 3 months. It is pulsatile and flat. Bulging may indicate increased ICP, but bulging while crying is normal. If the fontanel is depressed/sunken, it may indicate dehydration.
Abnormalities
- Craniosynostosis/Craniostenosis: premature closure of the fontanels and sutures. It can limit brain growth and available volume in the skull. This can increase ICP, result in mental retardation and brain herniation (through the foramen magnum), and death.
- Caput Succedaneum: swelling of the scalp caused by a prolonged second stage of labor. This swelling affects both sides, crossing the suture line. There is no special treatment required, as it normally resolves within two to three days.
- Cephalhematoma: collection or accumulation of blood caused by a rupture of periosteal capillaries. It does not cross the suture line, only affecting one hemisphere. It disappears in three to four weeks. The bleeding can contribute to jaundice.
- Craniotabes: localized softening of the cranial bones. It is commonly seen in early lightening and firstborn babies. It disappears in six weeks.
- Molding: overlapping cranial bones after birth as the bones squeeze together to pass through the birth canal. This is common among preterm babies.
Face Assessment
Observe the newborn’s face for symmetry. It should be primarily symmetrical, with only mild asymmetries. If the face is markedly asymmetrical, it can indicate facial nerve (CN VII) paralysis i.e. Bell’s Palsy.
- Eyes: pupils should be normally equal, round, reactive to light, and shows accommodation (PERRLA). The lacrimal duct in newborns is immature, so no tears form.
- Tests for blindness should done within the first ten days of life: Doll’s Eye Test (move head laterally; eyes should move in the opposite direction, indicating intact brain stem function) and Glabellar Tap Test (tap just above the nose; the eyes should blink as a protective reflex.
- Nose: the primary sign to look for from the nose is nasal flaring. Enlarged nares can indicate respiratory distress. This may be caused by obstruction by secretions, bone, or membranes (choanal atresia); or a lack of surfactant production.
Abnormalities
- Bell’s Palsy: paralysis of the facial nerve (CN VII). Management aims to prevent the premature disappearance of the sucking reflex. For this, the baby is fed with dropped and syringe, and encouraged to breastfeed. Aspiration precautions are applied.
- Coloboma: a keyhole appearance of the eyes from a hole formed in one of the structures of the eyes.
- Exotropia Strabismus: wall-eyed defect (◐‿◑) of the eyes, and Esotropia Strabismus: cross-eyed defect (◑‿◐) of the eyes. Non-physiologic strabismus is managed with occlusion therapy, surgery (SQUINT), corrective glasses, and laser therapy.
- Respiratory Distress Syndrome, A.K.A. Hyaline Membrane Disease. Its cause is unknown, but its predisposing factors include prematurity, LBW/SGA/LGA, and CS delivery.
- Craniofacial Defects: defects in the formation of the facial structures, such as a Cleft Lip/Palate deformity. Also found in Down Syndrome.
Respiratory Distress Syndrome
Also known as hyaline membrane disease, it is a disorder in the respiratory system of a newborn whose cause is unknown. Its predisposing factors include premature babies; LBW, SGA, and LGA babies; and CS delivery babies.
Management
- Continuous Positive Airway Pressure (CPAP): maintained positive airway pressure that keeps the alveoli open, preventing atelectasis.
- Oxygen Therapy: 40% FiO₂ (not higher, to prevent blindness/retrolental fibroplasia and emphysema/bronchopulmonary dysplasia).
- Incubation to provide a warm environment, allowing the newborn to conserve energy and avoid infection (via isolation). It is set at 34.4°C with a humidity of 55% to 65%. Minimal handling should be done, while utilizing touch therapy.
- Medication: steroids to promote surfactant maturation (Dexamethasone, Betamethasone), exogenous surfactant administration (Beractant) intratracheally, sodium bicarbonate for correction of acidosis, and gamma immunoglobulin.
- Monitoring: watch for anemia (BT, 50 mL PRBC), hyperbilirubinemia (Phototherapy, exchange transfusion), and malnutrition from excessive oxygen utilization, resulting in poor growth and development (TPN, Gavage, Breastfeeding)
Ear Assessment
The position of the ears is determined in relation to the line of the inner and outer canthus of the eye. Ears that are set lower than normal is abnormal, and can be seen in chromosomal defects such as Down syndrome, Patau syndrome, Edward disease; kidney defects; and craniofacial defects.
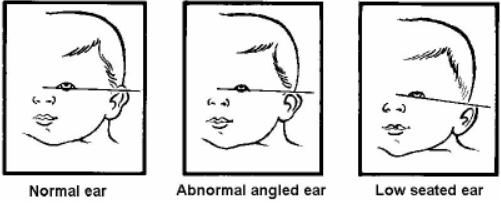
Abnormalities
- Down Syndrome: one of the characteristic appearances of Trisomy 21 is a low-set ear placement.
Down Syndrome
Trisomy 21, whose cause is unknown, but with the predisposing factors of advanced maternal age. Trisomy 21 indicates the presence of three 21st chromosomes, totaling 47 chromosomes rather than the expected 46. The expected lifespan of those with down syndrome is variable.
Assessment Findings
Down syndrome contains highly characteristic mental retardation and physical abnormalities, such as:
- Microcephaly
- Low-set ears
- Saddle nose
- Small mouth with macroglossia
- Short neck
- Short and stubby digits with single transverse line (simian crease)
- Hypotonic musculature
- Protuberant abdomen
- Small penis with cryptorchidism
Down syndrome commonly involves defects that can result in disability and dysfunction:
- Cardiac Defects: refer to Congenital Heart Defects
- Gastrointestinal Tract Defects: esophageal atresia
- Gastrourinary Tract Defects
Diagnostic Tests
- Alpha fetoprotein (AFP): a protein created by a growing liver, normally high in an unborn child. Testing of AFP presence in maternal serum can reveal low levels, indicating a potential genetic abnormality.
- Chorionic Villi Sampling: checks cells from the placenta (which are identical to cells from the fetus) to see if they have a chromosomal abnormality.
- Karyotyping: genetic testing to reveal the presence of extra or missing chromosomes.
Mouth Assessment
Observe the oral cavity for abnormal mucous membranes, secretions, odor, and appearance/structure. Craniofacial defects can also implicate the mouth, such as in the case of a Cleft Lip and Cleft Palate.
Abnormalities
- Epstein Pearls: white glistening epithelial cysts at the palate and gums, causing by extra loads of calcium.
- Teeth can be found if vitamin intake was excessive. They are extracted to prevent aspiration if they fall out, especially if neonatal reflexes have not yet developed.
- Vomiting can be projectile or non-projectile:
- Projectile: commonly caused by obstructive defects such as Pyloric Stenosis, Intussusception, and Aganglionic Megacolon.
- Non-projectile: commonly caused by non-obstructive defects such as infections, or chalasia (weakness of the lower esophageal sphincter)/GERD.
- Cleft Lip/Palate: a malformation of the lip/palate in a newborn. It is commonly genetic or due to maternal folic acid (Vitamin B₉) deficiency.
- Esophageal Atresia can be suspected by excessive drooling.
- Oral Moniliasis/Candidiasis: AKA oral thrush; caused by the bacteria Candida albicans, a fungus potentially acquired by the baby during passage through the birth canal. Management is mainly pharmacologic with antifungals (Nystatin) applied to the oral cavity with a gloved finger.
Cleft Lip and Cleft Palate
A malformation of the lip and/or palate. It is known to be genetic, or caused by maternal folic acid (B₉) deficiency. If uncorrected, it can result in feeding difficulties, speech defects, dental defects, altered normal body image, or even respiratory distress. The compromised tissues can also result in upper respiratory and ear infections (through the eustachian tube).
Surgical Management
A cheiloplasty or palatoplasty is used to reconstruct the closure of the unformed tissues. Nursing responsibilities during the preoperative phase include:
- Proper positioning during and after feeding: upright.
- Burp or bubble the newborn more often.
- Feeding utilizes cross-cut large holed nipple or Breck feeder technique.
- Monitor for complications: otitis media, etc.
- Orthodontic exercise and surgery for dental defects
- Speech therapy
During the postoperative phase, nursing care includes:
- Proper Positioning: prone if a cleft palate repair is done, and side-lying or supine if a cleft lip repair is done. An elbow restraint is used, released every two hours.
- Feeding with a rubber-tipped medicine dropper for cleft lip repari, and paper cups and soup spoon for cleft palate repair.
- Wound cleaning with hydrogen peroxide.
- Medications, including prophylactic antibiotics and analgesia.
- Avoiding incision disruption includes avoiding sucking (esp. for Logan’s Bar/Bow), suctioning, blowing, and any foreign object in the mouth.
Esophageal Atresia
It is the failure of the esophagus to form a continuous passage between the mouth and the stomach. It is a congenital defect often associated with other defects, warranting the assessment of other defects: (mn. VACTERL, VERT(i)CAL) vertebral, anorectal, cardiovascular, tracheal, esophageal, rectal, and limb defects. Its cause is unknown, but is also common in Down Syndrome.
- Diagnosis is done via x-ray.
Assessment Data
- Excessive drooling of saliva
- Choking after initial feeding
- Resistance upon NGT insertion
- Respiratory distress from secondary aspiration
- Abdominal distention: air trapping from tracheal fistulae connecting to the distal esophagus
- Cyanosis/tachypnea
- History of maternal polyhydramnios as the fetus does not swallow extra amniotic fluid. Approximately one-third of fetuses with esophageal atresia are found to have maternal polyhydramnios.
Medical Management
Surgery may be necessary depending on the type of defect. The creation of a gastrostomy or cervical esophagostomy can establish a patent esophagus. Fistulae are divided and the ends of the esophagus are anastomosed, before the gastrostomy is closed.
Nursing Management
Proper positioning (elevate head at 20° to 30°) is used to prevent aspiration. Regular suctioning is done.
- Administer oxygen if cyanosis occurs.
- Administer TPN as ordered (IV Hyperalimentation) as the primary source of nutrition.
Pyloric Stenosis
The narrowing of the pylorus/pyloric sphincter, which restricts gastric emptying and results in projectile vomiting in a newborn. It is the pyloric muscle that enlarges and thickens, decreasing the lumen of the pylorus. The stomach becomes a palpable olive-shaped mass and compensating peristalsis becomes visible. The child becomes irritable and restless.
- Surgery may be required to correct the stenosis. The Fredet Ramsted Procedure, a pyloromyotomy to separate hypertrophied pyloric muscle without any mucosal incision, releases the restriction of the pylorus. This is done with a laparoscope.
Nursing Management
- Feedings use thickened formula (rice cereal and milk) given through gavage.
- Prevent and correct dehydration from vomiting, with the potential use of IVF. Measure I&O.
- Monitor for complications such as metabolic alkalosis, acidosis, and dehydration.
Neck Assessment
The neck is often short in newborns. Notably, it is the site where the thyroid gland can be found. The nurse palpates for the thyroid gland, though it is normally not palpable in a newborn. A thyroid scan exposes T₃ (triiodothyronine) and T₄ (thyroxine) levels which, if low, diagnoses cretinism.
Abnormalities
- Cretinism: insufficient thyroid hormone production, potentially from iodine deficiency. This is one of the disorders tested in newborn screening as it can result in mental retardation. Treatment is hormone replacement therapy with Synthroid.
- Congenital Torticollis: “wry neck”; abnormal contractions of the sternocleidomastoid (SCM) muscle, resulting in twisting of the neck. It may be treated with exercise and the application of warm packs or compress, but in severe cases, a tenotomy may be done to release the contracted muscle.
Abdominal Assessment
The abdomen is normally dome-shaped and cylindrical. The stomach, at birth, is able to store ~90 mL, with rapid peristalsis (can commonly reverse as well) and short emptying time. Absorption is faster in the small intestines as they contain more secretory glands and a larger surface area. There are various palpable organs:
- Liver: 2 to 3 cm below the right costal margin.
- Kidneys: 1 to 2 cm above the umbilicus.
- Spleen: in the left upper quadrant.
Abnormalities
- Diaphragmatic Hernia: a herniation is the protrusion of an organ outside of its normal cavity. In the case of a diaphragmatic hernia, parts of the intestines can be pushed into the thorax, which can lead to the collapse of the lungs. It may be left-sided (Bochdalek Hernia) or right-sided (Morgagni Hernia).
- Omphalocele: the protrusion of abdominal organs through the umbilicus. The herniated organs are often contained by a sac, but is prone to infection. Until corrected, the nurse covers the defect with sterile saline dressing, preferably changed every two hours.
- Gastroschisis: the protrusion of abdominal organs through the abdomen via a defect or hole in the abdominal wall. The herniated organs are also very prone to infection (administer prophylactic antibiotics), and is reduced using a silastic silo (covering for the protruded organs, which reduces expulsion and maintains sterility until surgical repair is done.
- An overhead warming unit is used as the exposed organs increase heat loss.
- Intussusception is the telescoping self-insertion (invagination) of a section of the intestine. This is an obstructive disorder which results in pain (earliest sign) and projectile vomiting in newborns (bilous, fecaloid). These can form within three weeks of life.
- Palpation reveals a sausage-shaped mass in the abdomen, and the passage of currant jelly-like (bloody and mucoid) stool.
- Diagnosis reveals a coiled spring or staircase sign via a lower gastrointestinal series or barium enema.
- Treatment: barium enema, bowel milking, or if indicated, bowel resection with end-to-end anastomosis.
- Hirschsprung’s Disease/Aganglionic Megacolon: absent nerve cells (ganglions) in a section of the bowel which results in the loss of peristaltic movement.
Diaphragmatic Hernia
Parts of the intestines are pushed into the thorax, crowding the lungs, which can lead to lung collapse. It may be left-sided (Bochdalek Hernia) or right-sided (Morgagni Hernia).
Assessment Findings
- Dyspnea, tachypnea, and tachycardia from compromised lung volume.
- Cyanosis
- Asymmetrical chest development
- Concave abdomen from reduced volume, as some parts have herniated upwards.
- Asymptomatic cases can be found in those with a Morgagni Hernia, as the right lung has a higher capacity, as the left lung is already compensating the presence of the heart.
Medical Treatment
Diaphragmatic herniation is an indication for neonatal intensive care. The use of a mechanical ventilator may be required due to the compromised lung capacities, or even potentially an extracorporeal membrane oxygenator (ECMO). Surgery is used to correct the herniation.
Aganglionic Megacolon
Also known as Hirschsprung’s Disease, this is the absence of nerve cells (ganglions) in a section of the bowel results in the loss of peristaltic movement. Intestinal materials accumulate in the aganglionic section, and results in a megacolon/obstruction.
Assessment Findings
- Non-passage of meconium; passing of ribbon-like or pellet stool. Chronic constipation can be observed.
- Projectile vomiting
- Abdominal distention and shortness of breath
- Anorexia
Surgical Management
A colostomy is instated temporarily before a Swenson and Soave Procedure, followed by colostomy closure. Preoperatively, the nurse:
- Provides a daily enema (olive oil/diluted antibiotics for retention edema; NSS for non-retention edema).
- Feeds the child small, frequent low-residue feedings
- Measures abdominal circumference daily
- Elevates the head to ease breathing.
- Administer medications as ordered, e.g. stool softeners.
- Provides oral hygiene and psychosocial support
Anogenital Assessment
Assessment of the anus, rectum, perineum, and some parts of the genital system.
- The newborn is expected to pass meconium within 24 hours. Failure to do so can indicate obstruction (Imperforate Anus, Aganglionic Megacolon, Cystic Fibrosis)
- Stool varies with types: meconium (green-black, sticky, odorless, passed four times a day), transitional (yellow-green, slimy, six or more times a day), breast-fed baby stool (golden-yellow, mushy, soft, sweet odor, passed after feeding), and bottle-fed baby stool (pale-yellow, hard, formed, foul odor, passed once a day.)
Abnormalities
- Imperforate Anus: the non-formation of the anal passage, often with an associated fistula to the genitourinary system. It is suspected if the anal opening is observed to be absent or misplaced, if no stool has passed within 24 to 48 hours of life, or if stool has been passed elsewhere (vagina, base of the penis, scrotum, or urethra). Abdominal distention also results from the lack of stool passage. Surgery is used to correct the defect:
- Colostomy (Stage I) followed by a pull-through procedure (Stage II), followed by the closure of the colostomy (Stage III).
Urogenital Assessment
Assessment of the reproductive system, external genitalia, and internal structures (kidneys).
Abnormalities
- The newborn is expected to void six to eight times within the first 24 hours, and twelve to twenty times daily after. Failure to do so may suggest renal agenesis (non-formation of the kidneys), dehydration or an absence of the urinary meatus.
- Abnormalities of the meatus can result in altered direction or outflow of urine:
- Epispadias: the meatus opens atop the penis.
- Hypospadias: the meatus opens below the penis.
- Non-descent of the testes is checked with palpation of the scrotum. If the testicles are found to be undescended, a disorder known as cryptorchidism is determined. A complication of this condition is inguinal hernia, testicular cancer, and sterility.
- HCG and Testosterone as HRT can correct cryptorchidism. If necessary, surgical intervention (orchiopexy/orchidopexy) can be done to move the undescended testes into the scrotum.
- Transillumination of the scrotum can reveal clear fluid within the scrotum. This is intraabdominal fluid leaking into the scrotum, which is termed as a hydrocele. If a loop of intestines is also found within the scrotum, it is termed as a scrotal hernia.
- A hydrocele may be typed as non-communicating (closed off) or communicating (open; may result in herniation). The latter requires elective repair to prevent complication.
- Phimosis is present in most male babies. The foreskin is never retracted as it may cause laceration. This usually disappears within 6 months to 1 year, otherwise it will be treated with circumcision.
- Pseudomenstruation (false menstruation) may be observed in female babies, caused by the withdrawal of maternal hormones.
- Uric Crystals can be caused by urates, and appear as pink or brick-red crystals.
Extremity Assessment
The extremities are assessed for symmetry in movement, presence, and malformation. The movements of the extremities should be symmetrical, with asymmetry suggesting weakness or paralysis (commonly Erb’s palsy or Brachial Plexus Paralysis)
- The legs of a newborn are normally bow-like; bow-legged (varus) until toddlerhood.
- The feet of a newborn are flat due to fat deposits and usually point outward, but will be corrected (straight upward) by the time the child is ready to walk. The direction the foot points in and its general formation can be abnormal (Equinous, Varus, Valgus, Calcaneous)
Abnormalities
- Erb’s Palsy/Brachial Plexus Paralysis: damage to the brachial plexus nerve, which can limit the use of the arm. Damage commonly occurs during delivery. In the affected arm, the moro reflex is absent, the tonic neck reflex is incomplete, and there is decreased sensory and motor function. Infant reflexes are discussed here.
- The affected arm is abducted and rotated externally, then immobilized with a figure of eight or airplane splint.
- Amelia: absence of an entire limb
- Phocomelia: absence of arms or legs
- Hemimelia: absence of hands or feet
- Hip Dysplasia or Hip Dislocation tested with the Ortolani test, a test for hip dislocation (reduction). The examiner holds the hip and applies anterior pressure to flex the thigh on the hip, then rotates the thighs outward. If hip dysplasia is present, the head of the femur moves back into place. The Ortolani sign is a distinct click or clunk heard when the head of the femur slips forward in the acetabulum if hip dysplasia is present. The femur becomes displaced again once pressure is released.
- Casts and traction can be used to treat hip dysplasia/dislocation: hip spica cast, Bryant’s Traction.
- Equinous: downward-pointing toes (plantar flexion).
- Varus: inward-pointing toes (inversion).
- Valgus: outward-pointing toes (eversion).
- Calcaneus: upward-pointing toes (dorsiflexion).
- Equinovarus, a combination of equinous and varus malformations, is also known as Clubfoot, and is the most common defect. It is characterized by plantar flexion (downward-pointing toes) and inversion (inward-pointing toes). It may be managed with:
- Exercise
- Casting
- Arthrodesis
- Dennis Browne Application
Back Assessment
The back should be flat and straight. The lumbar curve only forms at six months of age.
Spina Bifida
Tufts of hair, dimples, or masses can indicate spina bifida. It is a deformity resulting from the incomplete closure of the vertebrae, allowing the protrusion and leakage of spinal components. This can also contribute to hydrocephaly.
- Spina Bifida Occulta: a hidden deformity, which can still be manifested by a tuft of hair, a dimple, or a small mass.
- Spina Bifida Cystica: a protrusion of the spinal contents through the open vertebral structure. This can be further classified according to the contents of the protrusion:
- Meningocele: protrusion of the meninges and cerebrospinal fluid.
- Myelomeningocele: protrusion of meninges, cerebrospinal fluid, nerve roots, and part of the spinal cord.
- Rachischisis: protrusion of the meninges and spinal cord.
- Hydrocephalus: an abnormal enlargement of the head from the retention of fluid within the cerebral ventricles.
Surgical Management
The sac is excised if possible. Surplus CSF is drained via a shunt to prevent ICP elevation. The shunt may be ventriculoperitoneal or ventriculoatrial. It is the duty of the nurse to observe for signs of shunt malfunction.
Nursing Management
The protruding sac should be protected from rupture. This can be done through positioning (prone with hips abducted and head slightly elevated), the application of a sterile donut ring around the sac, and covering the sac with sterile saline dressing changed every two hours.
Increased ICP related to hydrocephalus include characteristic physical manifestations:
- Macewen’s Sign a cracked pot sound
- Bossing Sign: a protruberant forehead
- Sunset: the iris is positioned lower than the sclera; the white of the eye is visible above the iris.
- Bulging and Tense Fontanels
- High Pitched and Shrill Crying
- Altered level of consciousness
- Changes in VS
Skin Assessment
The skin is assessed for its color and marks. The skin at birth is normally pink, potentially due to high RBC. Normal CBC values for an infant are:
- RBC: 4.4 to 7.5 million/mm³
- Hgb: 14.5 to 22.5 g/dL
- Hct: 45% to 65%
- WBC: 5,000 to 35,000/mm³
Abnormalities
- Cyanosis: blue/purple discoloration of the skin resulting from hypoxia.
- Pallor: paleness of the skin resulting from anemia, potentially due to ABO or Rh incompatibilities, hemolytic disorders, and bleeding. The decreased synthesis of Vitamin K can contribute to hemophilia, a deficiency or absence of clotting factors conducive to excessive bleeding. It may also be due to a deficiency in platelets.
- Jaundice: a yellow discoloration of the skin resulting from heightened levels of bilirubin. There are two types, physiologic (normal) and pathologic (abnormal):
| Parameter | Physiologic Jaundice | Pathologic Jaundice |
|---|---|---|
| Onset | 2nd day | 1st day |
| Duration | to 7 days (or to 14 for preterm babies) | More than 7 days (or more than 14 if preterm) |
| Bilirubin | 5 mg/day 12 mg/day on 3rd day | >5 mg/day 12 mg/day |
| Treatment | Sunlight and phototherapy | Phototherapy and exchange transfusion |
- Nevus Vasculosis (Strawberry marks) are elevated areas formed by immature capillaries and endothelial tissues.
- Nevus Flammeus (Portwine Stains): a macular purple or dark-red lesion or patches. It can be seen on the face, buttocks, thigh, and genitals.
- Tangiectasis Nevus: a flat, red area of capillary dilatation commonly seen at the glabella, upper eyelid, and upper lip.
- Mongolian Spot: a slate blue or gray patch caused by the accumulation of melanocytes commonly seen at the buttocks and back. This normally disappears by preschool age.
- Lanugo: fine downy hair seen at the back, upper arm, and shoulder. These are common in preterms as they often disappear by the eighth month of gestation. This disappears within two weeks.
- Vernix Caseosa: a white, cheesy substance seen all over the body that aids in delivery, thermoregulation, and acts as a bacteriostatic agent. This normally disappears within 24 hours.
- Erythema Toxicum: pink papules with superimposed vesicles seen on the face. This disappears in two weeks and is commonly the first rash of the newborn.
- Milia: small white spots commonly seen at the tip of the nose caused by clogged sebaceous glands. These are the whiteheads of the newborn.
- Desquamation: dry peeling-off of the skin