References:
- Wong’s Nursing Care of Infants and Children, 11th Edition, ISBN 978-0-323-54939-4, by Marilyn J. Hockenberry, David Wilson, and Cheryl C. Rodgers (Ch. 29, pp. 1349–1354)
Thalassemia is a common autosomal recessive genetic disorder worldwide. “Thalassa”, the root word of the disorder, means “sea,” and is applied to a variety of inherited blood disorders characterized by deficiencies in the rate of production of specific globin chains in hemoglobin. The name appropriately refers to people living near the Mediterranean Sea—namely, Italians, Greeks, and Syrians, or to their descendants which now has a wide geographic distribution as a result of genetic migration.
β-Thalassemia is the most common of the thalassemias and occurs in four forms: two heterozygous forms, one form which may either be homozygous or heterozygous, and a homozygous form. While recessive in nature, this gene can be expressed in various degrees:
- Thalassemia minor: generally an asymptomatic silent carrier state.
- Thalassemia trait: produces a mild microcytic anemia.
- Thalassemia intermedia: manifested as splenomegaly and moderate to severe anemia.
- Thalassemia major (Cooley anemia): results in a severe anemia that is not compatible with life without transfusion support.
Pathophysiology
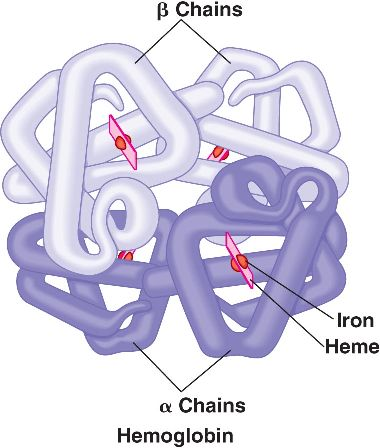
Hemoglobin is composed of four polypeptide chains—two alpha and two beta. In β-thalassemia, the beta chain proteins are inadequate, absent, or altered, resulting in the formation of defective hemoglobin.
- The unbalanced polypeptide unit is unstable and readily disintegrates, damaging the RBC, causing severe anemia. Because of this hemolytic process, the body compensates by producing more erythrocytes to the point of overabundance.
- Transfusion becomes necessary to suppress the bone marrow. However, excess iron from packed RBC transfusions and the hemolytic process can result in hemosiderosis, the deposition of iron in various organs.
Clinical Manifestations
The onset of clinical manifestations in thalassemia major may be insidious and not recognized until late infancy or early toddlerhood. The major consequences of thalassemia are caused by the pathologic condition, resultant chronic hypoxia, and iron overload from the supportive treatment of multiple blood supplements.
- Anemia results from the body’s inability to maintain a level of erythropoiesis commensurate with hemolysis. The bone marrow compensates by producing large numbers of immature cells, such as normoblasts and erythroblasts; large cells that are extremely thin and form bizarre shapes; and target cells, which have abnormal staining properties. As a result of the excessive production of abnormal RBCs, their life span is severely shortened.
- Progressive anemia eventually results in signs of chronic hypoxia—headache, irritability, precordial and bone pain, decreased exercise tolerance, listlessness, and anorexia.
- Splenomegaly occurs as a result of extramedullary hematopoiesis, rapid destruction of defective erythrocytes, and, rarely, progressive fibrosis from hemochromatosis. This may progress until the organ’s very size interferes with the function of other abdominal organs and respiratory expansion.
- “Hemosiderosis” refers to the deposition of iron in various tissues without associated tissue injury. “Hemochromatosis” refers to excess iron storage that results in cellular damage through unknown means.
- Retarded growth and delayed sexual maturation are common findings, but the exact reasons for them are unclear. Frequent epistaxis also occurs, but the exact reasons for it is also unknown. Hyperuricemia and gout from rapid cellular catabolism also occur.
Diagnostic Evaluation
Hematologic studies will show characteristic changes in RBCs show microcytosis, hypochromia, anisocytosis, poikilocytosis, target cells, and basophilic stippling of various stages. Hemoglobin electrophoresis distinguishes the variants of hemoglobin in the blood, and is confirmatory of the diagnosis. It finds elevated levels of HgbF and HgbA₂ because neither of these variants depend on the beta-chain polypeptides for synthesis.
Therapeutic Management
Supportive therapy is done to maintain sufficient hemoglobin levels to prevent bone marrow expansion and bony deformities and to provide sufficient RBCs to support growth and normal physical activity.
- Transfusions are the foundation of medical management, with a goal of maintaining the hemoglobin above 9.5 g/dl, an aim that may require transfusions as often as every 3 weeks.
- Iron Chelating Agents (Deferoxamine) are used to minimize the development of hemosiderosis and hemochromatosis. Deferoxamine (IV/subcut) or Deferasirox (oral) is given with oral supplements of vitamin C as it augments iron excretion in response to deferoxamine. This is administered over 8 to 10 hours (often during sleep) for 5 to 7 days a week. Adherence is highly important.
- Magnetic Resonance Imaging may be done to evaluate the iron content of the liver, heart, and other organs. This is the method of choice for guiding iron chelating therapy in combination with laboratory measurement of serum ferritin levels. A liver biopsy is used in some cases.
- Splenectomy may be necessary to decrease the disabling effects of abdominal pressure in severe splenomegaly. The spleen may also accelerate RBC destruction over time and therefore increase transfusion requirements.
Nursing Care Management
Basic to each of these goals is explaining to parents and older children the defect responsible for the disorder, its effect on RBCs, and the potential effects of untreated hemosiderosis.
- Promote compliance with transfusion and chelation therapy
- Assist the child in coping with the anxiety-provoking treatments and the effects of the illness.
- Foster the child’s and family’s adjustment to a chronic illness
- Observe for complications of multiple blood transfusions.