References:
- Wong’s Nursing Care of Infants and Children, 11th Edition, ISBN 978-0-323-54939-4, by Marilyn J. Hockenberry, David Wilson, and Cheryl C. Rodgers (pp. 1030–1043)
Anemia is a reduction in RBCs mass and/or hemoglobin concentration compared with normal values for age. The anemias are the most common hematologic disorders of infancy and childhood and are not diseases but manifestations of underlying pathologic processes. This may involve inadequate production of RBCs or its components, increased destruction of RBCs, and excessive loss of RBCs through hemorrhage.
Red Blood Cell Morphology
- Size (Cell Size): variation in red blood cell (RBC) sizes (anisocytosis)
- Normocytes (normal cell size)
- Microcytes (smaller than normal cell size)
- Macrocytes (larger than normal cell size)
- Shape (Cell Shape): variation in RBC shapes (poikilocytosis)
- Spherocytes (globular cells)
- Drepanocytes (sickle-shaped cells)
- Numerous other irregularly shaped cells
- Color (Cell Staining Characteristics): variation in hemoglobin concentration in the RBC
- Normochromic (sufficient or normal amount of hemoglobin per RBC)
- Hypochromic (reduced amount of hemoglobin per RBC)
- Hyperchromic (increased amount of hemoglobin per RBC)
The basic physiologic defect caused by anemia is a decrease in the oxygen-carrying capacity of blood and consequently a reduction in the amount of oxygen available to the cells. When the hemoglobin level falls sufficiently to produce clinical manifestations, the signs and symptoms (e.g., weakness, fatigue, and a waxy pallor in severe anemia) are due to tissue hypoxia. Cyanosis is usually not evident.
Signs and Symptoms of Anemia
Decreased Red Blood Cell Production
- Pallor
- Tachycardia
- Fatigue, headache
- Muscle weakness
- Systolic heart murmur
- Frontal bossing
Increased Red Blood Cell Destruction
- Icteric sclera, jaundice
- Fatigue, headache
- Tachycardia
- Dark urine
- Splenomegaly
- Hepatomegaly
- Low blood pressure (late sign of shock)
Increased Red Blood Cell Loss
- Pallor
- Fatigue, headache
- Muscle weakness
- Cool skin
- Tachycardia
- Decreased peripheral pulses
- Low blood pressure (late sign of shock)
Diagnostic Evaluation
In general, anemia may be suspected from findings on the history and physical examination, such as lack of energy, easy fatigability, and pallor. However, unless the anemia is severe, this may only become evident through alterations in the CBC (↓ Hgb, ↓ Hct, ↑ Reticulocytes).
- A peripheral smear may demonstrate significant changes in the shape of RBCs, such as sickled cells.
- Tests for measuring the amount of hemoglobin (mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration) in a single cell are helpful in determining the cause of the anemia.
- At times, a bone marrow aspiration may be necessary to evaluate the body’s ability to produce normal cells. For example, in leukemia the bone marrow is hyperplastic, whereas in aplastic anemia the bone marrow is hypoplastic or aplastic.
Therapeutic Management
The objective of medical management is to reverse the anemia by treating the underlying cause.
- In nutritional anemias the specific deficiency is corrected.
- In blood loss from acute hemorrhage, RBC transfusion may be given.
- In patients with severe anemia, supportive medical care may include oxygen therapy, bed rest, and replacement of intravascular volume with intravenous (IV) fluids.
Nursing Care Management
The nurse’s physical examination yields valuable evidence regarding the severity of the anemia and some indication of its possible cause. In interviewing the family, the nurse stresses the following areas: (1) nutrition, especially if the child is lactose intolerant or has inadequate intake of iron; (2) past history of chronic, recurrent infection; (3) eating habits, particularly pica (consumption of nonnutritive substances such as dirt, starch, lead-based paint chips, paper); (4) bowel habits and presence of frank blood in stools or black, tarry stools as a result of chronic blood loss; and (5) familial history of hereditary diseases, such as SCD or thalassemia.
- Prepare the Child and Family for Laboratory Tests. Several blood tests may be ordered sequentially. Multiple pricks may be required. Topical analgesics (EMLA, ELA-LMX) may allow better tolerance.
- Inform the family about the significance of the tests and why they are not done all at once.
- Encourage parents or another supportive person to be with the child during the procedure.
- Allow the child to play with the equipment on a doll or to participate in the actual procedure (e.g., holding the Band-Aid).
- Decrease Tissue Oxygen Needs: the nurse continuously assesses the child’s energy levels and minimizes tissue oxygen needs accordingly. In most instances of anemia this is not necessary, but when it is, the nurse must implement several important interventions (the same as those with a nursing diagnosis of fatigue or activity intolerance).
- Assess the child’s level of tolerance for activities of daily living and play, and make adjustments to allow as much self-care as possible without undue exertion. This allows the nurse to determine which activities will be physically taxing, and requires assistance as needed.
- Because dependency can be threatening, allow the child as much control of the environment as possible. For example, a child with severe anemia may be unable to walk to the bathroom but may be able to use a bedside commode or be transported in a wheelchair to the lavatory rather than having to use a bedpan.
- Scheduling activities throughout the day with planned rest periods in between maximizes the child’s energy potential without causing undue exertion.
- Anticipate and implement necessary safety measures (e.g., staying with the child when the child is out of bed and raising side rails when the child is in the bed to prevent falls).
- Plan diversional activities that promote rest but prevent boredom and withdrawal. Because short attention span, irritability, and restlessness are common in anemia and increase stress demands on the body, plan appropriate activities such as listening to music, watching television, reading or listening to stories, coloring or drawing, and playing board and card games.
- Choosing the appropriate roommate, such as a child of similar age with a diagnosis that also requires restricted activity, is another helpful intervention.
- If infants or young children are hospitalized, consider the importance of preventing separation from parents. Crying and fretfulness place greater stress demands on the body, which increase oxygen needs.
Signs of Exertion
Signs of exertion include tachycardia, palpitations, tachypnea, dyspnea, hyperpnea, dizziness, lightheadedness, diaphoresis, and change in skin color. The child looks fatigued (e.g., sagging, limp posture; slow, strained movements; inability to tolerate additional activity; difficulty sucking in infants).
Prevent Complications
- Infection: children with anemia are prone to infection because tissue hypoxia causes cellular dysfunction, and the disturbed metabolic processes weaken the host’s defenses against foreign agents. Infection also worsens the anemia by increasing metabolic needs and, in instances of chronic infection, also interferes with erythropoiesis and shortens the survival time of RBCs.
- Take all of the usual precautions to prevent infection, such as practicing thorough hand washing, selecting an appropriate room in a noninfectious area, restricting visitors or hospital personnel with active infection, and maintaining adequate nutrition.
- Blood Volume Depletion: drawing multiple blood samples may present a problem with cumulative blood loss and necessitate blood replacement. This situation occurs most often in infants with severe anemia. To prevent this situation, blood may be withdrawn through a continuous IV line and replaced after the exact amount needed has been tested and discarded. As a precaution, keep a record of the volume of blood withdrawn. Using micromethods of testing whenever possible minimizes the amount of blood required for the test.
- The nurse needs to observe for cumulative effects of blood loss, particularly signs of shock and increased hypoxia, and to explain to parents the necessity for taking multiple blood samples and the reason for blood replacement.
- Cardiac Decompensation: the main complication of anemia is cardiac decompensation, which can result from excessive demands on the heart due to increased metabolic needs or cardiac overload.
- The nurse should observe for signs and symptoms of heart failure such as tachycardia, dyspnea, rales, moist respirations, cough, and sweating. This is of first priority.
- Packed RBCs (rather than whole blood) are usually administered to prevent circulatory hypervolemia.
Iron Deficiency Anemia
Anemia caused by an inadequate supply or loss of iron is the most prevalent nutritional disorder worldwide and the most preventable mineral disturbance. While the prevalence of iron deficiency has been declining, it remains a relatively common health problem. Iron deficiency anemia can be caused by any number of factors that decrease the supply of iron, impair its absorption, increase the body’s need for iron, or affect the synthesis of hemoglobin. Management will be dependent on the cause of iron deficiency.
This discussion is limited to iron deficiency anemia resulting from inadequate iron in the diet.
Pathophysiology
Hemoglobin is the oxygen-carrying component of a red blood cell. Iron is required for the production of hemoglobin. One molecule of hemoglobin consists of protein (globin) combined with four molecules of a pigmented compound (heme). Each molecule of heme contains one atom of iron. If iron is deficient, hemoglobin levels decrease and the oxygen-carrying capacity of the blood is reduced.
Causes of Iron Deficiency Anemia
Inadequate Supply of Iron:
- Deficient dietary intake - Rapid growth rate - Excessive milk intake, delayed addition of solid foods - Poor general eating habits - Exclusive breastfeeding of infant after 6 months of age
- Inadequate iron stores at birth - Low birth weight, prematurity, multiple births - Severe iron deficiency in mother (hemoglobin level <9 g/dl) - Fetal blood loss at or before delivery
Impaired Iron Absorption
- Presence of iron inhibitors - Phytates, phosphates, or oxalates - Gastric alkalinity
- Malabsorption disorders - Lactose intolerance - Inflammatory bowel disease
- Chronic diarrhea
Blood Loss
- Acute or chronic hemorrhage
- Parasitic infestation
Excessive Demands for Iron Required for Growth
- Prematurity
- Adolescence
- Pregnancy
Clinical Manifestations
The clinical manifestations are directly attributable to the reduction in the amount of oxygen available to the tissues and resemble those seen in any type of anemia.
- Although infants with iron deficiency anemia tend to be underweight, many are overweight because of excessive milk ingestion (known as milk baby). These children become anemic for two reasons: milk, a poor source of iron, is given almost to the exclusion of solid foods, and increased fecal loss of blood occurs in 50% of iron deficient infants fed cow’s milk. Although chubby, these infants are pale (sometimes porcelain-like), usually demonstrate poor muscle development, and are prone to infection.
- Although the mechanism is unknown, iron deficiency anemia enhances the leakage of plasma proteins, which causes edema; retarded growth; and decreased serum concentration of the proteins albumin, gamma globulin, and transferrin (a protein that binds iron and transports it through the plasma).
- Other manifestations of iron deficiency include irritability, tachycardia, fatigue, glossitis, angular stomatitis, and koilonychia (concave or “spoon” fingernails).
Diagnostic Evaluation
- Laboratory Tests:
- RBC count may be normal, borderline, or moderately reduced. The problem lies in the concentration of hemoglobin, which is out of proportion with the normal number of erythrocytes.
- RBCs are typically small (microcytic), so that MCV is decreased. This may be diagnostic in infants if MCV reaches below 70 femtoliters (fl). The lower limit of normal of children 1 to 10 years old is 70 fl plus the child’s age in years.
- Reticulocytes remain normal or slightly reduced if iron stores are decreased. However, in severe anemia, tissue hypoxia elicits an erythropoietic response, elevating reticulocyte count by 3% to 4%.
- A stool analysis for occult blood (guaiac test) can rule out the possibility of chronic fecal blood loss, especially from milk intolerance or structural anomalies such as diverticulitis.
- Iron Studies
- Serum iron concentration (SIC) is normally about 70 mcg/dl in infants and slightly higher in older children. This test is performed in the morning, when SIC is highest.
- Total iron-binding capacity (TIBC) is the amount of transferring (iron-binding globulin), which is necessary for the transport of iron in the bloodstream. In an attempt to absorb more iron from exogenous sources, the body compensates by increasing TIBC. In iron deficiency anemia TIBC is elevated above the normal range of 350 mcg/dl (6 months to 2 years) or 450 mcg/dl (children >2 years and adults).
- Transferrin saturation is calculated by dividing the SIC by the TIBC and multiplying the result by 100 to express the value as a percentage. A transferrin saturation of 10% suggests anemia.
Therapeutic Management
Prevention is the primary goal and is achieved through optimum nutrition and appropriate iron supplementation. In infants, the following guidelines are recommended to prevent iron deficiency (American Academy of Pediatrics, Committee on Nutrition, 1999):
- Use only breast milk or iron-fortified formula for the first 12 months. Limit the amount of formula to no more than 1 L/day to encourage intake of iron-rich solid foods.
- Iron supplementation of 1 mg/kg/day should be provided by 4 to 6 months of age in full-term infants and 2 mg/kg/day by 2 months of age in preterm infants. Administer iron drops at a dosage of 2 to 3 mg/kg/day to a maximum of 15 mg/day of elemental iron to breastfed preterm infants after 2 months of age, and give iron-fortified infant cereal when solid foods are introduced.
- Universal screening for anemia should be performed at approximately 12 months of age with determination of Hgb concentration and assessment of risk factors associated with iron deficiency anemia.
After the diagnosis of iron deficiency anemia is made, therapeutic management focuses on increasing the amount of supplemental iron the child receives.
- Iron-fortified formula and/or inclusion of iron-rich food in the diet.
- Oral iron supplements are given if dietary inclusions of iron-rich foods is not provide sufficient supplemental quantities of the mineral. Iron supplements deliver ferrous iron, rather than ferric iron. Ferrous iron is more readily absorbed tan ferric iron and results in higher hemoglobin levels.
- Iron supplementation is prescribed as 3 to 6 mg of elemental iron per kilogram per day. Lower dosages are associated with fewer side effects. The dose is split into two or three doses between meals.
- Vitamin C appears to facilitate absorption of iron and may be given as vitamin C-enriched foods and juices with the iron preparation.
- Side effects are infrequent, especially in infants, but include nausea, gastric irritation, diarrhea or constipation, and anorexia. If vomiting and diarrhea occurs, the iron should be administered with meals and in gradually increasing doses.
- Hemoglobin levels rises from 0.1 to 0.4 g/dl/24 hr depending on anemia severity. A substantial increase should occur by the end of 1 month of oral therapy. If the hemoglobin level fails to rise after 1 month of oral therapy, it is important to assess for noncompliance, persistent bleeding, iron malabsorption, improper iron administration, or other causes of anemia.
- Parenteral IV/IM iron administration is effective, but painful, expensive, and have potential adverse effects (regional lymphadenopathy, transient arthralgias, serious allergic reactions). It is reserved for children with iron malabsorption, chronic hemoglobinuria, or intolerance to oral preparations.
- Deep IM iron administration is discouraged because it is painful and may leak into the subcutaneous tissue causing skin discoloration (hemosiderin staining) at the injection site.
- IV administration should be monitored carefully. A test dose is recommended before use.
- Transfusions are indicated for the most severe anemia and in cases of serious infection, cardiac dysfunction, or surgical emergency when anesthesia is required. Packed RBCs (2 to 3 ml/kg), not whole blood, are used to minimize the chance of circulatory overload.
- Supplemental oxygen is used when tissue hypoxia is severe.
Nursing Care Management
A primary nursing objective is to prevent nutritional anemia through family education. Nurses need to be aware of recommendations regarding iron supplementation during infancy and appropriate sources of dietary iron. The nurse should encourage parents to limit the quantity of milk, to use iron-fortified infant formulas, and to introduce solid foods. This may be difficult when parents believe milk is best for the infant and equate the resultant weight gain with “healthiness.” Although milk is an excellent food, it is deficient in iron, vitamin C, zinc, and fluoride.
Also stress that overweight is not synonymous with good health. If the infant has obvious signs of anemia, such as pallor, listlessness, frequent infections, and muscular weakness, point these out as evidence of suboptimum health. In some instances, it is helpful to chart the hemoglobin or hematocrit values to visually impress on parents the change in iron levels. Often, increased blood values correspond to improved physical status and reinforce the benefit of dietary or oral iron supplementation. It should be stressed with the family that the iron medication must continue for 2 to 3 months after blood values normalize to replenish the body’s iron stores.
Iron Administration
Ideally iron supplements are administered in two divided doses between meals, when the presence of free hydrochloric acid is greatest, and are accompanied by a citrus fruit or juice, which helps reduce iron to its most soluble state. The nurse must instruct parents regarding the proper administration of oral iron supplements.
- An adequate dosage of oral iron turns the stools a tarry green or black color. The nurse advises parents of this normally expected change and inquires about its occurrence on follow-up visits. Absence of greenish-black stool may be a clue to poor compliance.
- Oral iron supplementation in liquid form may stain the teeth. If possible, take the medication through a straw or give it through a syringe or medicine dropped toward the back of the mouth. Brushing after administration also lessens the discoloration.
- Iron ingested in excessive quantities is toxic. There should be no more than a 1-month supply in the home and it must be stored safely away from the reach of children.
Sickle Cell Anemia
Sickle cell anemia (SCA) is one of a group of diseases collectively termed hemoglobinopathies, in which normal adult hemoglobin (hemoglobin A [HgbA]) is partly or completely replaced by abnormal sickle hemoglobin (HgbS). SCD refers to a group of hereditary disorders, all of which are related to the presence of HgbS.
- SCA: the homozygous form of the disease (HgbSS), in which valine, an amino acid, is substituted for glutamic acid at the sixth position of the β chain. This is the most common form of the SCDs, and one of the most common genetic diseases worldwide, particularly of the African and African American populations.
- Sickle cell C disease: a heterozygous variant of SCD (HgbSC), characterized by the presence of both HgbS and hemoglobin C (HgbC), in which lysine is substituted for glutamic acid at the sixth position of the β chain.
- Sickle thalassemia disease: a combination of sickle cell trait and β-thalassemia trait. In the β+ (beta plus) form, some normal adult hemoglobin can still be produced. In the β0 (beta zero) form, there is no ability to produce normal adult hemoglobin.
Pathophysiology
The gene that determines the production of HgbS is situated on an autosome and is recessive. If both parents have sickle cell trait, there is a 25% chance with each pregnancy of producing an offspring with SCA.
The basic defect of the sickling phenomenon lies in the globin fraction of hemoglobin. It is composed of 574 amino acids. In HgbS, the molecular structure changes to form long, slender crystals that distort the cell membrane, so the cell changes from a pliable disk to a crescent- or sickle-shaped RBC. The filamentous forms are associated with much greater viscosity than those with the normal holly leaf structure of HgbA (when polymerized).
- Polymerization (alteration in structure) occurs in periods of dehydration, acidosis, hypoxia, and temperature elevation. In most instances, the sickling response is reversible under conditions of adequate oxygenation and hydration. During this time the RBCs are indistinguishable from normal erythrocytes on peripheral examination. However, repeated cycles of sickling and unsickling can cause the RBCs to become irreversibly sickled.
Although present at birth, sickling phenomenon is usually not apparent until later in infancy because of the presence of fetal hemoglobin (HgbF). These rapidly decrease within the first year.
Clinical Manifestations
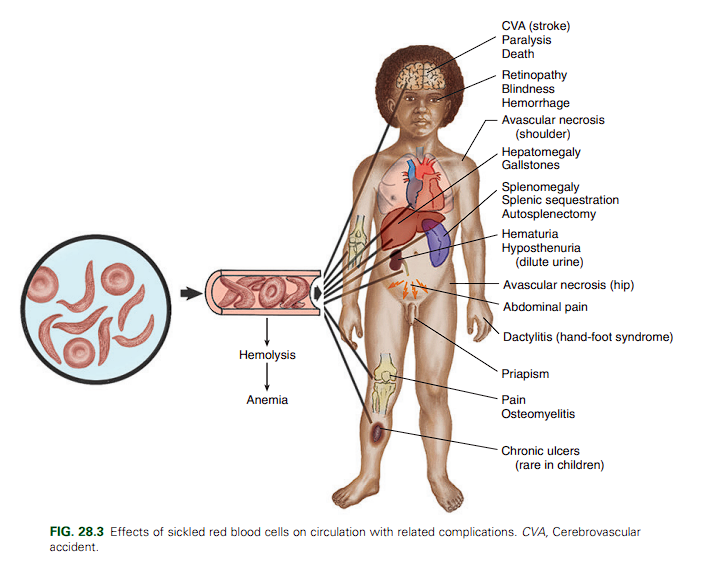
The clinical manifestations of SCA are primarily the result of (1) obstruction caused by the sickled RBCs, (2) vascular inflammation, and (3) increased RBC destruction. Sickled cells allow for abnormal adhesion, entanglement, and enmeshing of rigid sickle-shaped cells accompanied by the inflammatory process intermittently blocking microcirculation, causing vasoocclusion. The resultant absence of blood flow to adjacent tissues causes local hypoxia, which leads to tissue ischemia and infarction. Most of the complications seen in SCA can be traced to this process and its impact on various organs of the body.
Clinical Manifestations of Sickle Cell Anemia
General
- Possible growth retardation
- Chronic anemia (hemoglobin level of 6 to 9 g/dl)
- Possible delayed sexual maturation
- Marked susceptibility to sepsis
Vasoocclusive Crisis
- Pain in area(s) of involvement
- Manifestations related to ischemia of involved areas: - Extremities—Painful swelling of hands and feet (sickle cell dactylitis, or hand-foot syndrome), painful joints - Abdomen—Severe pain resembling acute surgical condition - Cerebrum—Stroke, visual disturbances - Chest—Symptoms resembling pneumonia, protracted episodes of pulmonary disease - Liver—Obstructive jaundice, hepatic coma - Kidney—Hematuria - Genitals—Priapism (painful penile erection)
Sequestration Crisis
- Pooling of large amounts of blood - Hepatomegaly - Splenomegaly - Circulatory collapse
Effects of Chronic Vasoocclusive Phenomena
- Heart—Cardiomegaly, systolic murmurs
- Lungs—Altered pulmonary function, susceptibility to infections, pulmonary insufficiency
- Kidneys—Inability to concentrate urine, enuresis, progressive renal failure
- Liver—Hepatomegaly, cirrhosis, intrahepatic cholestasis
- Spleen—Splenomegaly, susceptibility to infection, functional reduction in splenic activity progressing to autosplenectomy
- Eyes—Intraocular abnormalities with visual disturbances, sometimes progressive retinal detachment and blindness
- Extremities—Avascular necrosis of hip or shoulder; skeletal deformities, especially lordosis and kyphosis; chronic leg ulcers; susceptibility to osteomyelitis
- Central nervous system—Hemiparesis, seizures
Sickle Cell Crises
The most acute symptoms of the disease occur during periods of exacerbation called crises. There are several types of episodic crises: vasoocclusive, acute splenic sequestration, aplastic, hyperhemolytic, stroke, chest syndrome, and infection related.
- Vasoocclusive crisis (VOC), preferably called a painful episode or event is the most common type of non-life-threatening crisis. It is characterized by ischemia that causes mild to severe pain that may last from minutes to days. Pain may be localized or generalized, and is often migratory. Visceral hypoxia or gallstones may cause abdominal pain, priapism may occur, and arthralgia may be present. A low-grade fever may also be present.
- Sequestration crisis is caused by the pooling of large quantities of blood, usually in the spleen and infrequently in the liver, which causes a decrease in blood volume and ultimately shock. The splenic crisis may be acute or chronic. The chronic manifestation is termed hypersplenism. Resultant profound anemia and cardiovascular collapse may result in death.
- Aplastic crisis is diminished RBC production, usually triggered by infection with a virus (especially the human parvovirus) or other organism. When superimposed with the rapid destruction of RBCs, profound anemia results. Packed RBC transfusion is occasionally required in children exhibiting signs and symptoms of congestive heart failure.
- Hyperhemolytic crisis is an accelerated rate of RBC destruction characterized by anemia, jaundice, and reticulocytosis. This complication frequently suggests other coexisting conditions, such as viral illness; transfusion reactions to alloantibodies; or glucose-6-phosphate dehydrogenase (G6PD) deficiency, which is also common in African Americans.
- Stroke (cerebrovascular accident [CVA]) is a sudden and severe complication, often with no related illnesses. Sickled cells block the major blood vessels in the brain, which results in cerebral infarction causing variable degrees of neurologic impairment. Repeat CVA causes progressive brain damage in the majority of children who have already experienced one stroke and did not receive monthly transfusions because long-term red cell transfusions reduce the risk of CVA as supported by a moderate quality of evidence.
- Acute Chest Syndrome (ACS) is clinically similar to pneumonia. It is defined as a new pulmonary infiltrate on chest x-ray of a SCD patient that may be accompanied by chest pain, fever, cough, tachypnea, wheezing, and hypoxia. Researchers believe that a VOC or infection results in sickling in the small blood vessels of the lungs, with ensuing occlusion, stasis, and anemia. Repeated episodes of chest syndrome may cause restrictive lung disease and pulmonary hypertension.
Mortality in children with SCD
Overwhelming infection, especially with Streptococcus pneumoniae and H. influenzae type b as a result of defective splenic function, is the major cause of death in children with SCD under the age of 5 years. Repeated insults on the splenic sinuses by sickled cells result in impaired filtration and function, which allows the development of septicemia and possibly subsequent death.
Diagnostic Evaluation
Although SCA is usually reported during the neonatal period and early part of infancy, it may not be recognized until the toddler or preschool period during a crisis precipitated by an acute upper respiratory tract or GI infection.
- Screening: Sickledex— the sickle turbidity test is used. It can be performed on blood from a finger or heel stick and yields accurate results in 3 minutes.
- Confirmatory: Hemoglobin Electrophoresis is a test that “fingerprints” hemoglobin. It separates the sample into various hemoglobins through high-voltage electrophoresis and determines the types and percentages of each type. The test is accurate, rapid, and specific for detecting both the homozygous and heterozygous form of the disease.
Therapeutic Management
The aims of therapy are to prevent the sickling phenomenon, which is responsible for the pathologic sequelae, and to treat the medical emergency of sickle cell crisis. Medical management of a crisis is directed at supportive, symptomatic, and specific treatments. The main objectives are to provide:
- Bed rest to minimize energy expenditure and to improve oxygen utilization.
- Oxygen therapy has little therapeutic value unless the patient is hypoxic. It is usually not effective in reversing sickling or reducing pain because the oxygen is not able to reach the enmeshed sickled RBCs through the clogged vessels.
- Hydration through oral and IV therapy.
- Electrolyte replacement because hypoxia results in metabolic acidosis, which also promotes sickling.
- Analgesia for severe pain from vasoocclusion.
- Blood replacement to treat anemia and to reduce the viscosity of the sickled blood. It is used in aplastic, hyperhemolytic, and splenic sequestration crises; in stroke prevention; and before general surgery.
- Exchange transfusions (erythrocytapheresis) may be used to reduce the number of circulating sickle cells. This may be used in ACS, for high-risk surgeries, and after acute overt stroke to prevent recurrence and further tissue damage.
- Antibiotic therapy to treat any existing infection. Pneumococcal, H. influenzae type b, and meningococcal vaccines is recommended. Oral penicillin prophylaxis is recommended by 2 months of age to reduce the chance of pneumococcal sepsis.
- The nurse must help the family comply with medication regimen and seek medical attention immediately when the child has a fever of 38.5°C (101.3°F) or higher, an increase in spleen size, or severe pallor.
Nursing Care Management
Many nurses are involved in SCA screening programs to identify persons with the abnormal hemoglobin so that therapy can be implemented for homozygotes and genetic counseling provided for heterozygotes. During crises, the nurse assesses all systems that can be affected by circulatory obstruction.
- Minimize Tissue Deoxygenation: taking frequent rest breaks during physical activities, avoiding contact sports if the spleen is enlarged because rupture will cause massive internal hemorrhage, and avoiding environments with low oxygen concentration (high altitudes, nonpressurized airplane flights), and avoiding known sources of infection. Even mild infections should warrant medical attention at once.
- Promote Hydration: the nurse emphasizes the importance of adequate hydration to prevent sickling and delay the vasoocclusion and hypoxia-ischemia cycle.
- Minimum daily fluid intake for children is approximately 1600 ml/m²/day. Specific instructions are required for parents, rather than a simple “force fluids” or “encourage drinking”.
- Dilute urine is not a valid sign of adequate hydration, because the kidney’s ability to concentrate urine is impaired. Other indications of fluid loss, such as dry mucous membranes, dry diapers, weight loss, and a sunken fontanel should be noted.
- Enuresis is expected as a result of increased fluid intake and impaired kidney function. Normal interventions such as limiting fluids at night are not therapeutic.
- Minimize Crises: the nurse stresses to parents the importance of adequate nutrition, frequent medical supervision, proper hand washing, and isolation from known sources of infection. Keep in mind that children also need to live a normal life. Overprotection can be as devastating emotionally as an infection is physically. Parents need to be aware of the need to seek prompt medical care at the first sign of any infection.
- In splenic sequestration, gently measure the size of the spleen because increasing splenomegaly is an ominous sign. Conversely, a decrease in the size of the spleen denotes response to therapy.
- Promote Supportive Therapies:
- Analgesic therapy is a difficult problem. Undermedication is a common problem, which can result in false pretenses of drug addiction when the patient requests for additional doses sooner than expected. Various analgesics may need to be tested with varying schedules before adequate relief is achieved.
- The nurse should combine any pain management program with psychologic support to help the child deal with the depression, anxiety, and fear that accompany the disease. This includes regular visits with the child to discuss his or her concerns during the hospitalization and positive reinforcement of adaptive coping skills.
- Frequently, heat to the affected area is soothing. Cold compresses are not applied to the area because doing so enhances vasoconstriction and occlusion.
- Bed rest is usually well tolerated during a crisis, although the actual rest obtained depends a great deal on pain alleviation and the use of organized schedules of nursing care. Although the objective of bed rest is to minimize oxygen consumption, some activity, particularly passive range-of-motion exercises, is beneficial to promote circulation. Usually the best course is to let children determine their activity tolerance.
- Blood Transfusions: the nurse has the responsibility of observing for signs of transfusion reaction, especially signs of cardiac failure from hypervolemia.
- Decrease Surgical Risks: the main surgical risk is hypoxia from anesthesia. However, emotional stress, the demands of wound healing, and infection potentially increase the sickling phenomenon, both in children with the disease and in those with the trait. The primary nursing objectives are to minimize each of these threats preoperatively and postoperatively by keeping the child well hydrated, preparing the child psychologically, and preventing infection.
Quality Patient Outcomes for Sickle Cell Disease
- Early recognition of signs and symptoms of sickle cell anemia
- Tissue deoxygenation minimized
- Sickle cell crisis prevented or quickly managed
- Pain appropriately managed
- Stroke prevented
- Prophylactic penicillin regimen followed
- Hypoxia prevented when surgery is necessary
- Pneumococcal, H. influenzae type b, and meningococcal vaccines administered
Aplastic Anemia
Aplastic anemia (AA) is a rare and life-threatening disorder that affects approximately 2 to 6 in 1 million children and adults each year.
- AA refers to a condition in which production of the formed elements of the blood is simultaneously depressed. The peripheral blood smear demonstrates cytopenia, including at least two of the triad that consists of leukopenia, thrombocytopenia, and profound anemia.
- Hypoplastic anemia is characterized by a profound depression of RBC formation but normal or slightly decreased production of WBCs and platelets. Pure RBC aplasia is a congenital conditions in which all erythroid series cells are almost or completely absent.
AA can be primary (congenital, or present at birth) or secondary (acquired). The best-known congenital disorder of which AA is an outstanding feature is Fanconi anemia syndrome, a rare hereditary disorder that is characterized by pancytopenia, hypoplasia of the bone marrow, and patchy brown discoloration of the skin due to the deposition of melanin. Acquired AA may be caused by:
- Human parvovirus infection, hepatitis, or overwhelming infection
- Irradiation
- Immune disorders such as eosinophilic fasciitis and hypoimmunoglobulinemia
- Drugs such as certain chemotherapeutic agents, anticonvulsants, and antibiotics
- Industrial and household chemicals, including benzene and its derivatives, which are found in petroleum products, dyes, paint remover, shellac, and lacquers
- Infiltration and replacement of myeloid elements, such as in leukemia or the lymphomas
- Idiopathic (in most cases no identifiable precipitating cause found)
Diagnostic Evaluation
Clinical manifestations, which include anemia, leukopenia, and decreased platelet count, is usually insidious, not unlike that seen in leukemia. A definitive diagnosis is determined from bone marrow examination, which demonstrates the conversion of red bone marrow to yellow, fatty bone marrow.
Therapeutic Management
The objectives of treatment are based on the recognition that the underlying disease process is failure of the bone marrow to carry out its hematopoietic functions. Therefore therapy is directed at restoring function to the marrow and involves two main approaches:
- Immunosuppressive therapy (IST) to counter the presumed immunologic responses that prolong aplasia. This is used in children with acquired AA who do not have a matched sibling bone marrow donor. It consists of
- Antilymphocyte globulin (ALG) or antithymocyte globulin (ATG) are the principal drug treatment used for AA with a presumed cause of autoimmunity. ATG is administered intravenously for 12 to 16 hours for 4 days after a test dose to check for hypersensitivity.
- Oral cyclosporine (T cell-dependent autoimmune response suppressant) is given for several weeks to months.
- Replacement of the bone marrow through transplantation.
- Although IST is effective in ameliorating the pancytopenia of aplastic anemia, IST has a significant risk in development of somatic mutations that may evolve into myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML). Recent studies have suggested that HLA-matched unrelated bone marrow should be strongly considered for children with acquired severe AA who lack an HLA-identical sibling donor.
- Hematopoietic Stem Cell Transfer (HSCT) should be considered early in the course of the disease if a compatible donor can be found. Transplantation is more successful if performed before multiple transfusions have sensitized the child to leukocytes and HLAs.
- With IST and HLA-identical sibling donor, the allogenic bone marrow transplantation offers 90% chance of long-term survival. Recently, survival outcomes from unrelated HLA-identical bone marrow transplantation are approaching the success seen with HLA-identical sibling bone marrow transplant.
Nursing Care Management
The care of the child with AA is similar to the care of the child with leukemia and includes preparing the child and family for the diagnostic and therapeutic procedures, preventing complications from the severe pancytopenia, and emotionally supporting the family in the face of potentially fatal outcome.