References:
- Wong’s Nursing Care of Infants and Children, 11th Edition, ISBN 978-0-323-54939-4, by Marilyn J. Hockenberry, David Wilson, and Cheryl C. Rodgers (Ch. 26, pp. 1074–1075)
- TRA Modules
Hirschsprung’s disease (HD) is a congenital anomaly that results in mechanical obstruction from inadequate motility of part of the intestine.
- It accounts for about one fourth of all cases of neonatal intestinal obstruction (1 in 5000 live births).
- Four times more common in males than females
- Etiology is unknown; may be familial in a few cases.
- Mutations in the RET protooncogene is involved in 17% to 38% of children with short-segment HD and in 70% to 80% with long-segment involvement.
- Short-segment HD aganglionosis is restricted to the internal sphincter, rectum, and a few centimeters of the sigmoid colon.
- Long-segment HD aganglionosis can affect the entire colon, part of the small intestine, or the entire intestinal tract (ileocecal valve to the anus); total colonic aganglionosis (2% to 13% of cases).
Pathophysiology
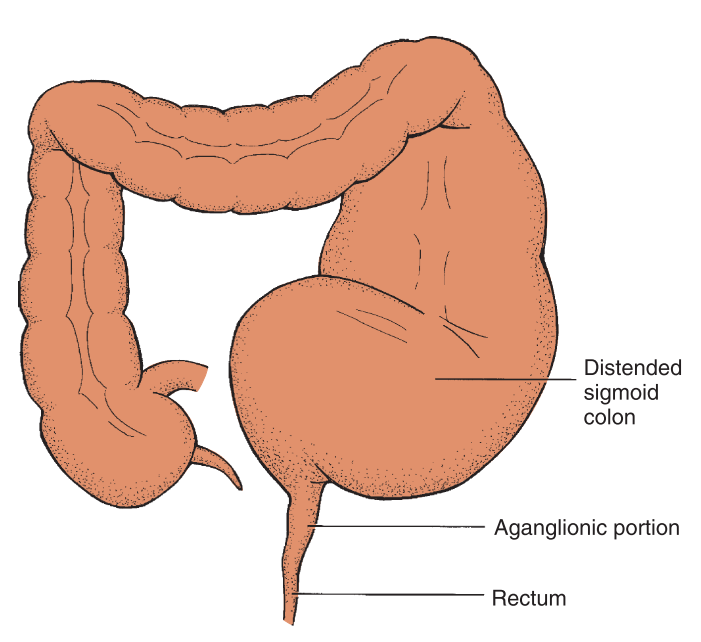
Aganglionosis of the intestine results in a loss of the rectosphincteric reflex and an abnormal microenvironment of the cells of the affected intestine. The term congenital aganglionic megacolon describes the primary defect, which is the absence of ganglion cells. The reduction of enteric nervous system stimulation inhibits the internal sphincter’s ability to relax. Because ganglion cells are parasympathetic, sympathetic stimulation of the intestine remains unopposed, resulting in increased intestinal tone.
Clinical Manifestations
Most children with HD are diagnosed in the first few months of life. Depending on the age when symptoms appear, manifestations may vary.
- Newborn Period:
- Failure to pass meconium within 24 to 48 hours. Typically, 99% of term infants will pass meconium within the first 48 hours of life, while less than 10% of infants with HD will do so.
- Refusal to feed
- Bilious vomiting
- Abdominal distention
- Infancy:
- Failure to thrive
- Constipation
- Abdominal distention
- Episodes of diarrhea and vomiting
- Signs of enterocolitis, a complication (explosive, watery diarrhea; fever; appears significantly ill)
- Childhood:
- Constipation
- Ribbonlike, foul-smelling stools
- Abdominal distention
- Visible peristalsis
- Easily palpable fecal mass
- Undernourished, anemic appearance
Diagnostic Evaluation
In neonates, the diagnosis is initially suspected on the basis of clinical signs of intestinal obstruction or failure to pass meconium. In infants and children the history is an important part of diagnosis and typically includes a chronic pattern of constipation.
- Rectal Examination: absence of feces and tightness of internal sphincter. Leakage of liquid stool and accumulated gas may occur upon examination if the affected segment is short.
- Contrast Enema: visualization of the transition point between the aganglionic distal segment and the dilated proximal colon (megacolon). However, this may only become evident in children two months of age or older.
- Rectal Biopsy: a biopsy is confirmatory once it determines the absence of ganglion cells from a full-thickness or suction biopsy.
- Rectal Manometry: a balloon catheter is inflated in the rectum to apply pressure to the rectal wall and attempt to stimulate the internal anal sphincter. It measures pressure applied by the internal and external sphincter in response to the balloon. A normal finding will show a relaxation of the internal sphincter followed by a contraction of the external sphincter. In HD the external sphincter contracts normally but the internal sphincter fails to relax.
Therapeutic Management
Most children with HD require one of three operative procedures once the child is stabilized with fluid and electrolyte replacement and colonic cleansing (enemas).
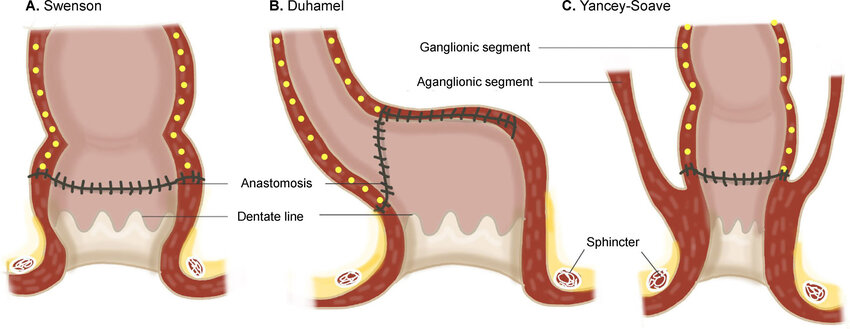
- Soave pull-through procedure
- Swenson procedure
- Duhamel procedure
Nursing Care Management
Nursing care will depend on the age of the child and type of treatment being used.
- Neonate: the primary focus is to aid the parents adjust to a congenital defect in their child, foster infant-parent bonding, prepare the parents for medical-surgical intervention, and prepare the parents to assume the care of the child after surgery.
- Monitoring for complications: the most serious complication of HD is enterocolitis. Preoperative care may include decreasing bacterial flora with oral or systemic antibiotics and colonic irrigations using antibiotic solution. In neonates, this may not be necessary as the bowels are still sterile.
- Postoperative Care: involve and aid the parents of the child in caring for the child. Allow them to help with feedings and observe for signs of wound infection, irregular passage of stool (constipation or true incontinence), or the development of complications.
- Measure abdominal girth daily and as necessary.
- Assess the surgical site for redness, swelling, and drainage.
- Assess the stoma (if a colostomy was done) for bleeding or skin breakdown. The stoma should be pink-red and moist.
- Maintain NPO status until bowel sounds return or flatus is passed, usually within 48 to 72 hours.
- Maintain the nasogastric tube to allow intermittent suction until peristalsis returns.
- Maintain IV fluids until the child tolerates appropriate oral intake, advancing the diet from clear liquids to regular as tolerated and as prescribed.
- Preventing Complications: anal strictures are a potential problem after surgery. Daily dilatations may be required, which can be done by parents who can be taught to perform care at home.
- Colostomy Care: a diverting colostomy may be used in some cases of HD. Parents are taught about the use and care of colostomy appliances.